
|
Development of novel and selective small molecule inhibitors of AeKir1 ion channel for use as first-in-class mosquitocides.
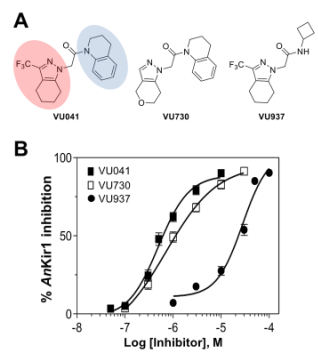
Mosquitos transmit numerous pathogens that cause emerging and re-emerging diseases that cause significant public health concerns world-wide. Most recently, the World Health Organization has deemed the emerging Zika situation as a “public health emergency of international concern”, with an alarmingly high number of reported cases of microcephaly and Guillain-Barré syndrome. Unfortunately, only a limited number of insecticides are available which has led to the evolution of resistance against these agents. Work in my collaborators laboratories have shown that targeting the Kir1 ion channel would be a novel target for developing mosquitocides with a novel target of action. Starting from a high-throughput screen, my laboratory has discovered a number of Kir inhibitors which are active only after injection. More recently, we have discovered VU041, a topically active AeKir1 inhibitor that elicits kidney failure and death in mosquitos, along with my collaborators Dr. Jerod Denton (VU) and Dr. Peter Piermarini (OSU). Further work is on-going to develop more potent and selective Kir inhibitors. |
|
|
Development of novel, potent and selective activators of GIRK1/2
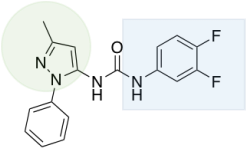
G protein-gated Inwardly-Rectifying K+ (GIRK) channels are critical mediators of cell excitability in the brain and heart. GIRK channels have gained significant interest as a potential therapeutic target for multiple medical indications; however, the lack of subtype-selective GIRK channel activators has precluded rigorous characterization of the physiological relevance and therapeutic potential of these channels. Our interest in this project is to develop potent and selective GIRK1/2 channel activators as potential therapies for mood-related disorders, epilepsy, and nociception/analgesia. Starting from a high-throughput screen, a novel pyrazole based urea analog (ML297) was discovered as a selective activator. However, ML297 is limited in use due to poor DMPK properties and low brain penetration. On-going work will be concentrated on looking at urea replacements as well as work around the pyrazole moiety. Advanced compounds will be evaluated for their in vitro potency and selectivity (Dr. David Weaver, Vanderbilt University) and then in an in vivo animal model of stress-induced hyperthermia (Dr. Kevin Wickmann, University of Minnesota), a preliminary mouse model of anxiety. This work is being funded by an R01 (MH107399) through the NIMH. |
|
Identification of novel TRPC5 inhibitors that protect kidney filter damage
Along with collaborators from Harvard University (Dr. Anna Greka), my laboratory recently identified a small molecule that inhibits TRPC5 and protects the kidney, in vivo, from filter barrier damage. The compound, ML204, was discovered from an HTS through the MLPCN network. Albuminuria, which the accumulation of plasma albumin into the urine is caused by a defective filtration in the kidney and is often a consequence of cardiovascular or metabolic diseases and there are no current treatments. This work has identified a specific strategy for the development of a novel therapeutic for this condition. This work is being funded by an R01 (DK103658) through the NIDDK.
Along with collaborators from Harvard University (Dr. Anna Greka), my laboratory recently identified a small molecule that inhibits TRPC5 and protects the kidney, in vivo, from filter barrier damage. The compound, ML204, was discovered from an HTS through the MLPCN network. Albuminuria, which the accumulation of plasma albumin into the urine is caused by a defective filtration in the kidney and is often a consequence of cardiovascular or metabolic diseases and there are no current treatments. This work has identified a specific strategy for the development of a novel therapeutic for this condition. This work is being funded by an R01 (DK103658) through the NIDDK.
|
Optimization of dopamine D4 antagonists for the treatment of L-DOPA-induced dyskinesias (LIDs)
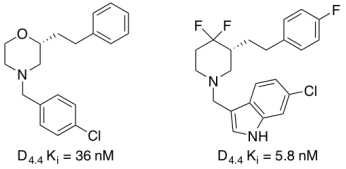
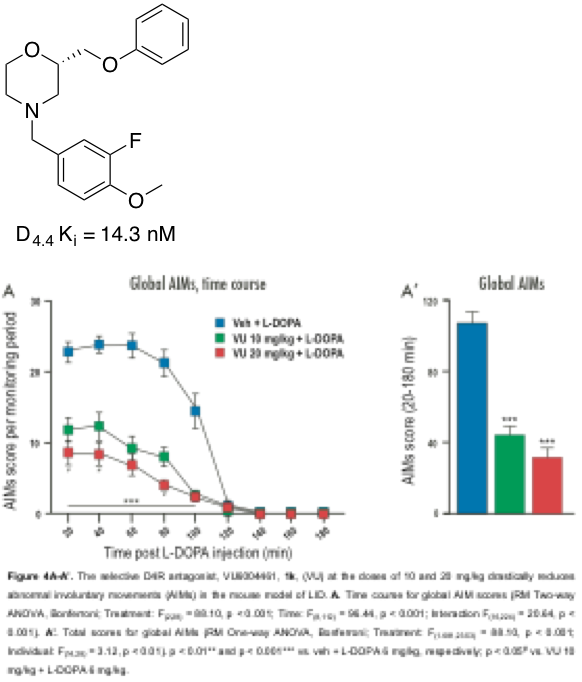
The dopamine receptors are members of the Class A G-protein coupled receptors (GPCRs) superfamily. The dopamine receptors are further subdivided into five subtypes, D1-5, which are further divided into two distinct families (D1-like and D2-like). The D1-like family consists of the D1 and D5 receptors and activate adenylyl cyclase whereas the D2-like family consist of the D2, D3 and D4 receptors and inhibit adenylyl cyclase. There is high homlogy between the dopamine receptors with D1 and D5 ~80% identical and the D2, D3, and D4 receptors having ~75% similarity. This high homology has been difficult to overcome in the discovery of selective D4 ligands. Recent studies indicate a role for the dopamine D4 receptor subtype in the modulation of BG circuitry underlying the pathophysiology of LIDs in PD. D4 receptors are present within the basal ganglia, namely within the striatum, globus pallidus, and subthalmic nucleus. Due to the distribution, the D4 receptor could play a major role in basal ganglia signaling and motor dysfunction. A number of D4 antagonists have been shown to be effective in alleviating L-DOPA-induced dyskinesias in both the 6-OHDA and MPTP-lesioned nonhuman primate models. My laboratory has developed numerous scaffolds that have shown excellent potency and selectivity as dopamine receptor 4 antagonists (chiral morpholine, chiral oxymethyl morpholine, and chiral difluoropiperidine based scaffolds). Evaluation of the lead compound in a mouse model of PD-LIDs (Dr. Angela Cenci, Lund University, Sweden) showed drastic reduction of global AIMs score in a dose-dependent manner. Further work is on-going to develop a novel, orally bioavailable antagonists. This work was funded by a Therapeutic Pipeline grant through the Michael J. Fox Foundation for Parkinson's Research. |
|
